Louis

Posts: 6436
Joined: Jan. 2006
|
| Quote (Tracy P. Hamilton @ Sep. 25 2008,00:31) | | Quote (Henry J @ Sep. 24 2008,11:04) | | Quote (Tracy P. Hamilton @ Sep. 24 2008,09:19) | � | Quote (Lou FCD @ Sep. 20 2008,13:18) | 4. Phospate Group
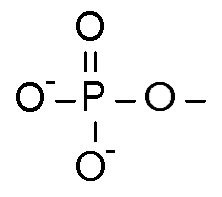
A Phosphate Group is a Phosphorus bonded with two negatively charged Oxygen atoms, one regular Oxygen atom, and double bonded with one Oxygen atom. It's molecular formula is PO<sub>4</sub><sup>2-</sup> or sometimes OPO<sub>3</sub><sup>2-</sup>, to separate the double bonded Oxygen.
Here is where the lecture ended. Although I asked in a later lecture about the odd bondings here that seem to break the rules that we earlier set forth, Doc basically said, "It's complicated, and you don't need to know that for this class, though you'll learn about it in a Chemistry class if you take one." Ok, fair enough. |
Chemical education discourages the use of the double bond in phosphate, and in sulfate groups as well. �As you noticed, it violates the octet rule, and does so unnecessarily! �There are some molecules where we chemists violate the rule out of necessity, such as having five atoms bonded to P, which requires 5 bonds.
The double bonds in phosphate and sulfate are historical, hence the biologists still use it a lot because that is how they learned it. �:O |
So is the proper way to do it to put a - sign on three of the O's and a + sign on the P? |
YES. �Don't listen to the old fogey Louis.
What Louis says is correct, though. �
Chemists have this resonance idea, but the question is which is more important: �a structure that obeys the octet rule but has +1 on P and -1 on each O, or a structure that disobeys the octet rule and has charge 0 on P, and 0 on one O. �Is having a positive charge on P so bad, and having negative on O so bad? �Answer: no. �Louis finds it offensive, apparently. �:p
I wonder if Louis would draw BF3 with a double bond or not, and whether there is any pi bonding in it (his justification for drawing P=O in phosphate). |
BF3?
LOL Sure you could draw a double bond, or a B+ (as you could for phospohrous/sulfur, I'll get to the key difference in a moment), although in this case it's a pi orbital formed from a combination of a lone pair in the F 2p orbital and the empty 2p orbital of the sp2 hybridised boron. The same sort of resonance picture applies here, as that double bond is delocalised over the three bonds. But again, there is definite pi character, the hallmark of a "double" bond (the quotes indicate that thee static formalisms are inadequate to explain a resonance picture)
Spectroscopically the B-F bond in uncomplexed BF3 is slightly shorter than calculated. Also, BF3 is trigonal planar (derived from it's sp2 hybridisation), contrast this with the comparable nitrogen halide or even ammonia (trigonal pyramidal molecules which get their shape from the tetrahedral arrangement of filled orbitals (bonding and non-bonding) around the central N atom). Also contrast it with the Lewis acidity of the boron halides, BBr3 is a much stronger Lewis acid than BF3, as demonstrated by its comparative ease of hydrolysis, precisely because that pi bonding is less strong due to the comparatively poor overlap between the smaller boron 2p orbital and the larger higher energy 4p orbital. The trend in Lewis acidity of the boron halides goes AGAINST that which we would expect from the halide electronegativity trend precisely because of this. BF3 has "less" of a "naked" B+ than does BBr3 by that reckoning, again though it is an oversimplification to treat these molecules as either double bond or + and -, reality, as ever, is more subtle. However, I'd be "happier" to have a double bond in BF3 than BBr3, and "happier" to have a B+ in BBr3 than BF3.
As I mentioned above (and I don't find +s on P/S/B/anything offensive btw, I love my +s, I'll even go as far as radical anions and carbenes (shock horror)! Hence why I mentioned ylides to Henry) these diagrammatic formalisms are just that: formalisms, as is the octet rule. They are simplified ways of looking at a much more complex picture. Switching between them in appropriate circumstances is a useful tool in developing mechanistic insight.
In all seriousness, if I've given any other impression (i.e. that one should stick a double bond in bloody everywhere) then I apologise, I was being amusing! Every bond can be thought of as a + and - under the right circumstances, just as it can be thought of as a pair of radicals for example. Most of the time (unless for example detailed mechanistic work has been done with the usual array of kinetics, radical/triplet quenches etc) these diagrammatic formalisms are interchangeable.
The major difference between P/S and B is a) their size, and b) the availability of those d orbitals. That makes a huge difference. In the case of phosphorous and sulfur there are plenty of examples of restricted rotation/bond shortening around the supposed "double" bond. For example the whole basis of the difference in mechanism between the Wittig reaction and the Horner-Wadsworth-Emmons reaction is based on tuning ylide electronic states so that rotation around the P-C bond axis is restricted/unrestricted, and hence why one gets different (ratios of) geometric isomers out the other end of the reaction. Those electronic states can be so tuned that there is even debate about whether the HWE involves the same type of phosphorous species as the Wittig.
The key is to look at the data, formalisms are all well and good, but the data is king! The spectroscopic data for phosphate (for example) show something more than a P-O sigma bond. Depending on what you are doing (IR, UV etc) you see that bond a slightly different way. Hence why, for mechanistic and diagrammatic purposes, one uses these interchangeable formalisms to simplify the resonance (i.e. quantum mechanical) picture.
Now a really good question is how one would draw diborane (B2H6, because if BF3 and phosphate anions blow people's minds about bonding, then diborane (or even higher boranes) is like the LSD version!
Cheers!
Louis
--------------
Bye.
|






















