niiicholas
Posts: 319
Joined: May 2002
|
From:
http://www.iscid.org/ubbcgi....#000058
=====
Just a short note, in the unlikely event that others were following the details of this thread. �Earlier in this thread Nelson expressed severe doubts about Blocker et al's (2003) model of Spa47/FliI as a hexamer, corresponding to the hexamer shape of the F1 subunit of F1F0-ATPase:
�| Quote |
Nic, there is no such thing as a protein complex of Spa47. It's ficticious, imaginary, like Santa Clause, the Easter Bunny, Pumpkin Head, the Matrix, X-men, spider-man, super-man, echo and the bunnymen(actually they exist). It's made up. Of course it fits, the whole thing is imaginary. How can it be a confirmed prediction? Once the post-doc finds it (or something like it) then the prediction that the model makes can be confirmed.
|
(this was important as a hexamer of Spa47/FliI would be yet another similarity between these ATPases (Spa47 in a T3SS, FliI in a flagellum) and the F1 ATPase subunit)
Anyhow, I just came across a brand-new article which would appear to empirically confirm the hexamer model:
�| Quote |
Mol Microbiol. 2003 Jun;48(5):1349-55. �
Oligomerization and activation of the FliI ATPase central to bacterial flagellum assembly.
Claret L, Calder SR, Higgins M, Hughes C.
Cambridge University Department of Pathology, Tennis Court Road, Cambridge CB2 1QP, UK.
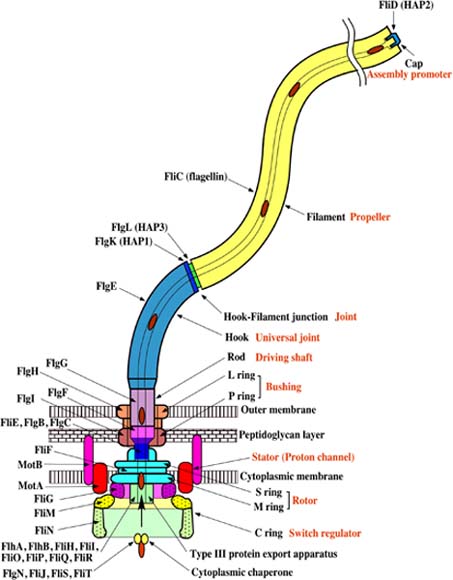
FliI is the peripheral membrane ATPase pivotal to the type III protein export mechanism underlying the assembly of the bacterial flagellum. Gel filtration and multiangle light scattering showed that purified soluble native FliI protein was in a monomeric state but, in the presence of ATP, FliI showed a propensity to oligomerize. Electron microscopy revealed that FliI assembles to a ring structure, the yield of which was increased by the presence of a non-hydrolysable ATP analogue. Single particle analysis of the resulting electron micrograph images, to which no symmetry was applied, showed that the FliI ring structure has sixfold symmetry and an external diameter of approximately 10 nm. The oligomeric ring has a central cavity of 2.5-3.0 nm, which is comparable to the known diameter of the flagellar export channel into which export substrates feed. Enzymatic activity of the FliI ATPase showed positive co-operativity, establishing that oligomerization and enzyme activity are coupled. Escherichia coli phospholipids increased enzyme co-operativity, and in vitro cross-linking demonstrated that they promoted FliI multimerization. The data reveal central facets of the structure and action of the flagellar assembly ATPase and, by extension, the homologous ATPases of virulence-related type III export systems.
|
Here is their graphic:
�
http://groups.yahoo.com/group/aefiles/files/FliI_ATPase_image.gif
From their conclusion:
�| Quote |
A previous report has indicated that His-tagged FliI can exist in monomeric form (Minamino and Macnab, 2000a), and our gel filtration and multiangle light scattering analysis of the native soluble enzyme confirmed this earlier work; all FliI was recovered as the monomeric form. However, when we incubated the enzyme with its substrate ATP, it was evident that FliI has a tendency to oligomerize. Electron microscopy and two-dimensional image processing of FliI stabilized by the non-hydrolysable ATP analogue AMP-PNP revealed that the ATPase assembles to ring structures that exhibit a strong sixfold symmetry around a central cavity, which may represent a central channel. The data, achieved without the application of any symmetry, indicate that FliI is possibly a homohexamer. Such a structure would be comparable to that of the ATPases of the distinct type IV protein export pathway in Helicobacter pylori and E. coli, ascertained by comparable means (Krause et al., 2000a,b). Our data also revealed that ATPase activity of FliI displays positive co�operativity, indicating interactions between subunits, i.e. FliI oligomerization is coupled to activation of the enzyme.
|
=====
Also, for the transport thread:
http://www.antievolution.org/cgi-bin....12;t=11
| Quote |
Mol Microbiol. 2003 Jun;48(5):1145-56. �
Tracing pathways of transport protein evolution.
Saier MH.
Division of Biological Sciences, University of California at San Diego, La Jolla, CA 92093-0116, USA.
We have conducted bioinformatic analyses of integral membrane transport proteins belonging to dozens of families. These families rarely include proteins that function in a capacity other than transport. Many transporters have arisen by intragenic duplication, triplication and quadruplication events, in which the numbers of transmembrane alpha-helical hydrophobic segments (TMSs) have increased. The elements multiplied may encode two, three, four, five, six, 10 or 12 TMSs and gave rise to proteins with four, six, seven, eight, nine, 10, 12, 20, 24 and 30 TMSs. Gene fusion, splicing, deletion and insertion events have also contributed to protein topological diversity. Amino acid substitutions have allowed membrane-embedded domains to become hydrophilic domains and vice versa. Some evidence suggests that amino acid substitutions occurring over evolutionary time may in some cases have drastically altered protein topology. The results summarized in this microreview establish the independent origins of many transporter families and allow postulation of the specific pathways taken for their appearance.
|
One relevant bit:
| Quote | | Evolution of small integral membrane transporters from one, two or three TMS precursors As noted above, many channel proteins/peptides exhibit just one or two TMSs (Fig. 1A). We currently have no proof of an intragenic duplication event allowing a primordial single TMS protein or peptide to give rise to a two TMS protein. However, one family of two TMS proteins, the bacterial type III target cell pore (III TCP) family (TC #1.C.36), gave comparison scores in excess of 9 SD (20� 30% identity; 65�80% similarity) when the first and second TMSs were compared (X. Zhou and M. H. Saier, Jr., unpublished results). Because the regions compared were short ( < 30 residues), we do not consider that these values establish homology (Saier, 1994; 2000a). This illustrates the difficulty in evaluating statistical significance for comparisons made between very short protein sequences. Our failure to establish homology in such cases probably does not reflect the absence or rarity of such duplications during evolutionary history. |
...and...
| Quote |
An example of a three TMS�four TMS interconversion exists in the Mot/Exb superfamily (TC #1.A.30; Cascales et al., 2001; Table 3). MotAB serves as a proton channel that allows bacterial flagellar rotation to be coupled to the proton motive force (pmf) by catalysing transmembrane proton flux. The MotA and MotB proteins together comprise the H+ channel. MotA is a four TMS protein, whereas MotB is a one TMS protein. Homologues of the MotA�B pair include the ExbB�D pair, which together are believed to provide the proton channel that allows the pmf to drive active transport across the outer membrane of the Gramnegative bacterial envelope (Cascales et al., 2001). The three TMS ExbB protein is homologous to the four TMS MotA protein, whereas the one TMS ExbD protein is homologous to MotB. The N-terminal TMS of MotA is lacking in ExbB. Although it is impossible to know whether a three or four TMS protein was the precursor of these homologues, it is clear that three and four TMS homologues have arisen by the gain or loss of an N-terminal TMS.
|
...and...
| Quote |
The several dozen examples that have been cited in this review provide an emerging picture of how the majority of transmembrane transport proteins probably evolved. The primordial systems were simple one, two or three TMS peptides that could form flexible oligomeric channels. These provided simple transmembrane diffusion functions with low degrees of substrate selectivity. A need for higher degrees of specificity, for coupling two or more transport processes (i.e. co-transport, antiport) and for energy coupling for solute accumulation or expulsion provided the driving force for carrier evolution. The formation of stereospecific binding sites, strict stoichiometric recognition and conformational coupling either within the transporter polypeptide chain itself or with other superimposed energy-coupling subunits were all required.
Such requirements resulted, first, in the evolutionary appearance of more constrained channels that could assume carrier functions, secondly, in the appearance of larger, more complex obligatory secondary carriers that could no longer catalyse passive diffusion and, thirdly, in the emergence of primary active transporters and group translocators with superimposed energy-coupling subunits. Thus, the ancestral precursors of all these transporter types were simple peptide channels. The pathways most frequently taken were evidently tandem intragenic duplications giving rise to larger helical bundles that had the potential to form discrete stereospecific intramembranous substrate binding sites. They could also be constrained for coupling to other transport processes and, through conformational coupling, they were subject to control by a superimposed primary energy-yielding process such as ATP hydrolysis. We assume that none of these requirements could be satisfied by the ancestral one, two or three TMS polypeptides that served as the ancestral precursor as no such carrier has yet been documented in the scientific literature. Thus, the primary advantages provided by intragenic duplication or triplication to generate large transmembrane proteins over oligomerization of smaller peptides were (i) specificity; (ii) stoichiometric transport coupling; and (iii) control by energy expenditure. These last two processes allow accumulation of specific solutes within or expulsion of solutes from a cell, processes that simple channels are incapable of catalysing.
Experimental confirmation of this proposal will require a combination of bioinformatic and molecular biological approaches. The functional consequences of the proposed structural constraints are likely to prove more difficult to ascertain than the structural constraints them-selves. Future studies are likely to shed light on such structure�function relationships, particularly as three-dimensional structures of more transport proteins become available. They should also allow more extensive documentation of the evolutionary pathways identified here and reveal new types of evolutionary processes not yet recognized. Multiple approaches should allow definition of the requirements for flexibility and rigidity in the construction and evolutionary modification of transport proteins.
|
And speaking of that, we go to supplementary material and we have:
http://www-biology.ucsd.edu/~msaier/supmat
Notably, this paper:
| Quote |
Peabody, C. 2003, "Type II Protein Secretion and its Relationship to Bacterial Type IV Pilli and Archael Flagella"
...which apparently has not yet been published but which surely is going to be important.
|
One more paper:
| Quote |
Biochim Biophys Acta. 2002 May 3;1562(1-2):6-31.
Protein-translocating outer membrane porins of Gram-negative bacteria.
Yen MR, Peabody CR, Partovi SM, Zhai Y, Tseng YH, Saier MH.
Division of Biology 0116, 9500 Gilman Drive, University of California at San Diego, La Jolla, CA 92093-0116, USA.
Five families of outer membrane porins that function in protein secretion in Gram-negative bacteria are currently recognized. In this report, these five porin families are analyzed from structural and phylogenetic standpoints. They are the fimbrial usher protein (FUP), outer membrane factor (OMF), autotransporter (AT), two-partner secretion (TPS) and outer membrane secretin (Secretin) families. All members of these families in the current databases were identified, and all full-length homologues were multiply aligned for structural and phylogenetic analyses. The organismal distribution of homologues in each family proved to be unique with some families being restricted to proteobacteria and others being widespread in other bacterial kingdoms as well as eukaryotes. The compositions of and size differences between subfamilies provide evidence for specific orthologous relationships, which agree with available functional information and intra-subfamily phylogeny. The results reveal that horizontal transfer of genes encoding these proteins between phylogenetically distant organisms has been exceptionally rare although transfer within select bacterial kingdoms may have occurred. The resultant in silico analyses are correlated with available experimental evidence to formulate models relevant to the structures and evolutionary origins of these proteins.
|
Edited by niiicholas on June 12 2003,03:35
|